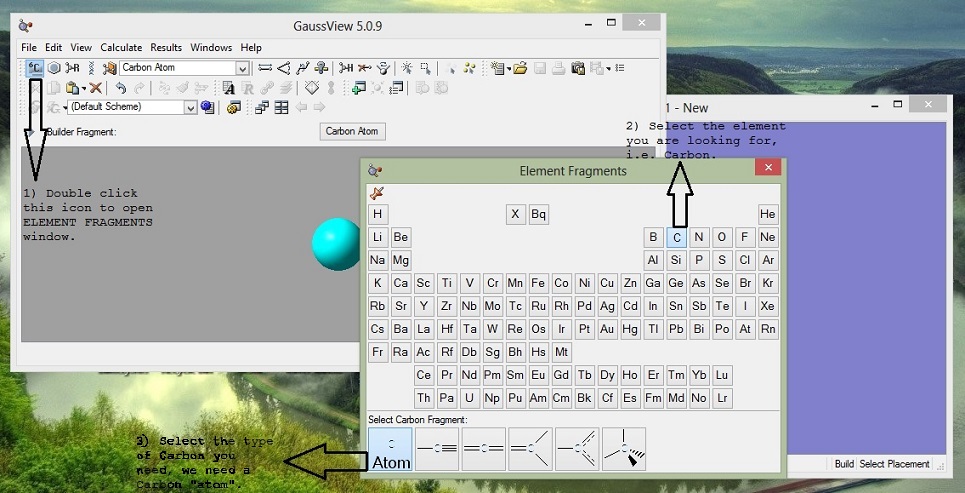
2. Add Atoms
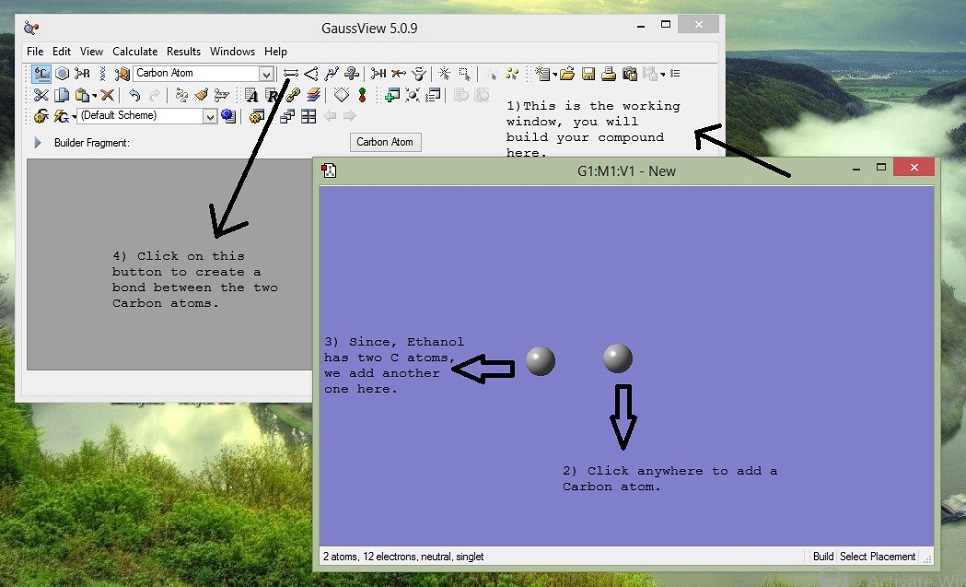
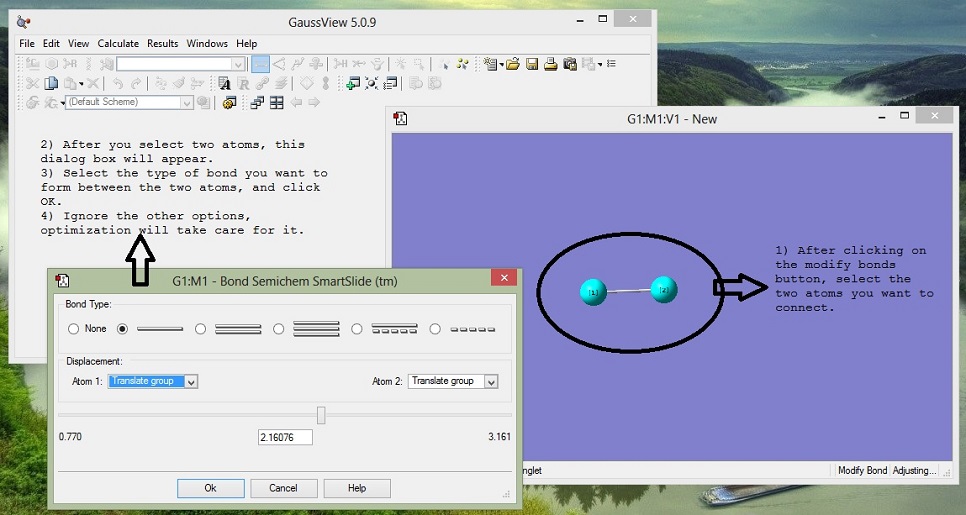
3. Join Atoms
4. Build Molecule
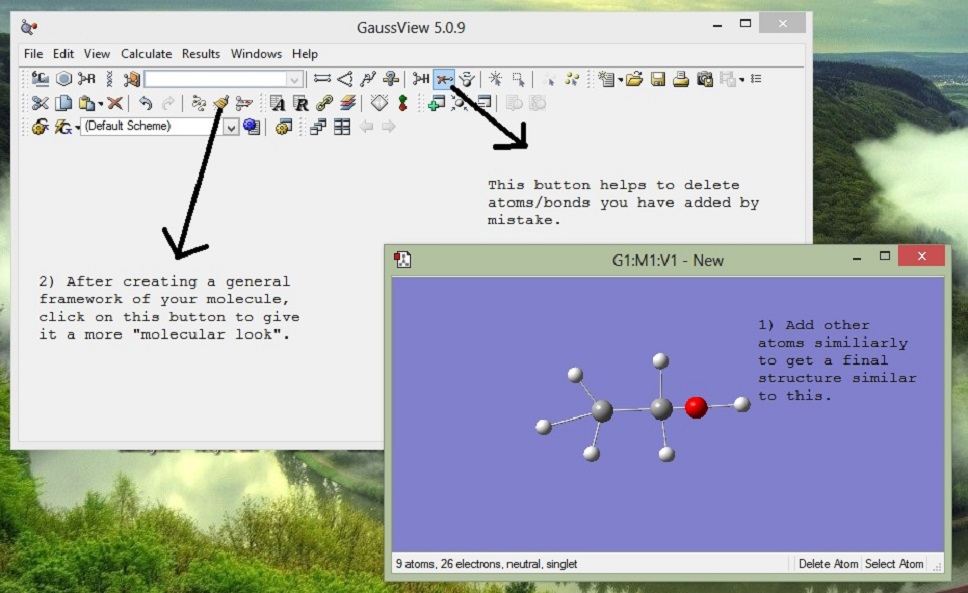
5. Brush
A pdb file contains the x,y,z coordinates of different atoms in the system. A system is basically a collection of different types of molecules located within the boundary of a box. Why is it called pdb or protein data bank? Well, because during the 1980's people started to keep the coordinates of different crystal structures of protein. These structures are highly useful (the recent noble prize in Chemistry was given for obtaining these protein structures, yeah it's that important!!) and so the scientists decided to keep a global database of all these structures. Thus the name Protein data bank. These structures became so ubiquitous that soon the structures of all the molecules, say polymers, carbohydrates, etc, were stored in these form. Though the word stored is not really a good way of saying it, but you get the idea, right? Here is a sample pdb file in case you haven't seen one till date.
If you had a look in the file above, you might have notices that it contains a lot of information. To be honest, none of it is required in our simulations. All we need are the three coordinates, which begins at about 1300th line of the file. Other information are relevant for citing the source of the pdb file used, how the pdb file was obtained, how the crystal structure was obtained, who obtained it, where, so on and so forth. In our simulations, we will be finding the thermodynamic property of ethanol. There are a number of ways in which people obtain these pdb files.
RCSB : the official pdb repository, highly unlikely you won't find your compound here
Gaussian : small molecules are often optimized in this quantum software
Published articles: some papers provide the pdb structures of the compound studied in their work, check out the supporting info.
Other sources: there are many, good luck <3
We will be using Gaussian. Firstly, ethanol is a rather small molecule and it will hardly take time to optimize the structure of the molecule in Gaussian. Secondly, it's often hard to get the native structure of a compound in the data bank, they are always in the form of some complex, which might not always be a good choice for carrying out simulations.
Gaussian is a quantum chemical calculation software. GaussView is a side kit to help you provide a graphical interface
for specifying the calculation parameters in Gaussian. Though you will run it, get the pdb file and forget it, I would ask you to know a bit about it too.
Gaussian was developed by the people at CMU ("THE CMU" where Soumya Di went), specifically by John Pople. He got a noble prize for his contributions
to the field of quantum chemistry. In your first year quantum course, you might have been told about the EY=HY equation (and the time dependent one too),
and that it is really hard to get the solution for many electron molecules. Well, this software tries to do the hard work by
figuring out a solution for any number of electron. There is a whole world of quantum thoery going on behind the screen, from the likes of plank, einstein, schrodinger, pauli,
feynman, pople, fokker, this softwares tries to apply all those learnings to help you get the pdb. Gaussian tries to find the
best possible orientations of the atoms in the molecule to minimize the energy. To be honest it does much more than that. I wish I could explain all that.
While on the one hand Gaussian is famous for having provided the world with an easy to use quantum calculation software; on the other hand it is a proprietory software
(meaning IITG pays a lot of money each year so that you can play and laugh at it). Further, Gaussian has also been in controvery for having
banned it's father
(Dr. Pople, the Noble Laureaute) from using the software (what the ****?). I think thats a lot of information for today.
1. Start GaussView 5.0
2. Add Atoms
3. Join Atoms
4. Build Molecule
5. Brush
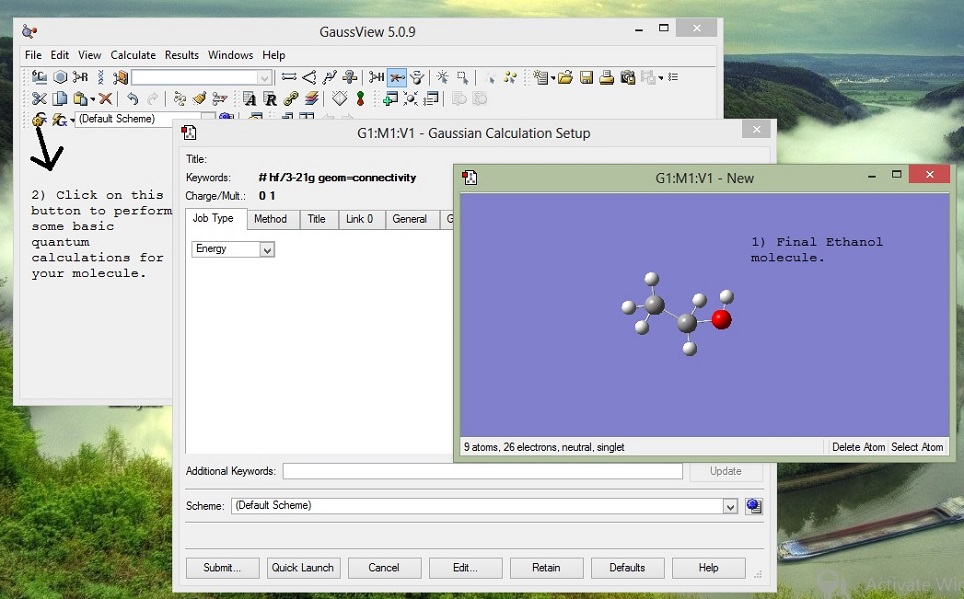
GaussView helped us to build the a general model of the molecule. But that structure we obtained is not reliable. It is not the most energy efficient structure per se. Next, we perform quantum calculations to optimize the structure of our molecule and to get the partial charges for the molecule. There are many ways to perform the calculations, depending on different molecule and system you may need to use different options of basis sets,etc. Its always lethal to carry out work you don't understand. Here is a simple introductory note on the working of abinitio methods.
1. continued from last pic..
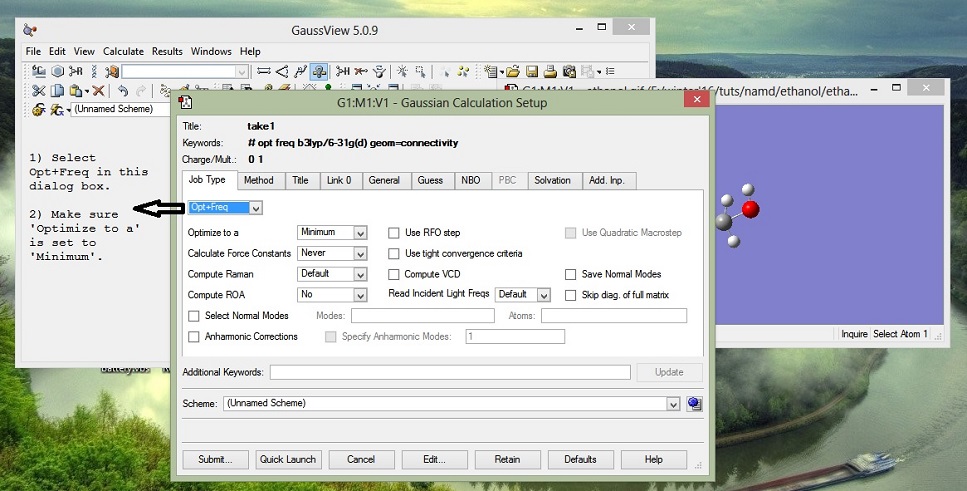
2. DFT by Walter Kohn
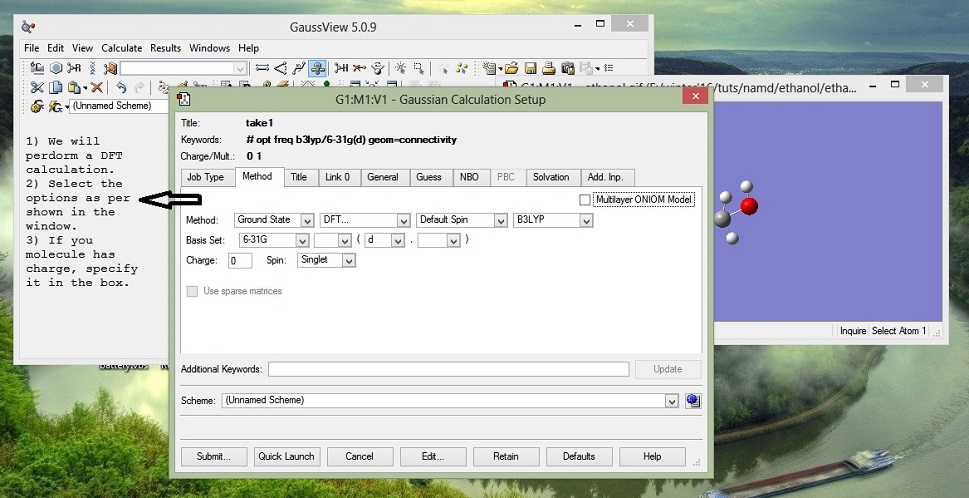
3. Submit
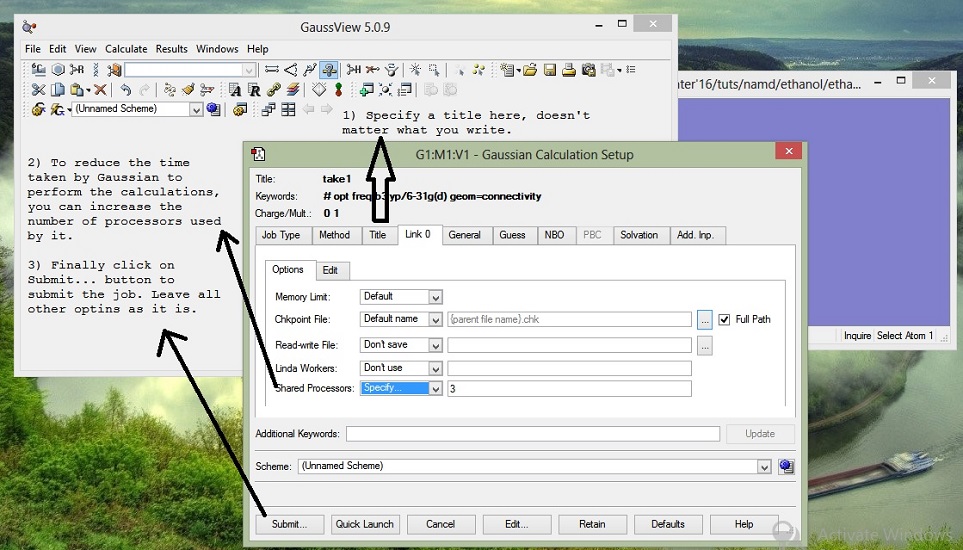
4. Extra comments:
In the Additional keywords section (see above pic, at the bottom), you can add: pop=chelpg
This will give you charges based on calculations...? write more
After clicking on submit.. button, save the Gaussian input file in a separate folder. The calculations will start and go on for some time depending on your molecule and computer specifications. This task should take around 4-5 minutes. After the job is over, follow these steps:
The pdb we obtained is rather ill formated (have a look at the 1YA4 pdb). We need to reformat it to add resname, resid, etc. These will help in easy visualization later in the process. It's essential to reformat the pdb hereon, else you might find it really difficult later on. Here is a tcl script to reformat the pdb. VMD has the option to execute tcl scripts. Follow these steps:
Previous Page Main Page Next Page